AI设计的分子能合成吗?合成可及性评分全解析|7大模型横评
如果你花了一整周让 AI 生成百万个漂亮的新分子,第二天收到一封邮件问:“里头有多少个能实际做出来?”
你盯着屏幕,大概不太想回答。
这种沉默我们懂。
AI 画分子式确实快,但实验室里的瓶瓶罐罐并不认 AI 画的饼。合成可及性这五个字,卡住了很多 AI 制药项目的脖子。
为了解决这个问题,合成可及性评分(Synthetic Accessibility Score,SA Score),成了核心工具。
下面我们就聊聊目前领域里 7 个主流的合成可及性预测模型,说说它们的技术原理、好用的地方,还有各自适合的场景。
现在行业里做合成可及性预测,主要走三条技术路线。
第一条是靠规则和经验的启发式方法。靠专家的化学知识和化学经验规则,用人为定好的评分函数,去判断一个分子的合成难度。
第二条是机器学习的数据驱动方法。拿海量的化学反应数据喂给模型,通过统计学习或者深度学习模型,自己摸透合成的规律。
第三条是基于逆合成的方法。直接模拟真实的合成路径,用逆合成分析技术,倒推这个分子到底能不能做出来。
以下,我们对这些代表性方法进行逐一详解。
SA Score:靠 “眼熟”打分
2009 年,诺华制药研究院的一群人琢磨出 SA Score,它的核心逻辑很简单,就是从两个维度给分子的合成难度打分。
一个是分子本身的复杂度。算分子量、有多少个环系统、多少个立体中心这些基础的拓扑特征,直接反映分子结构本身有多复杂。
另一个是片段的熟悉度。把 PubChem 数据库里 100 万个分子,按标准规则拆成子结构片段,再查每个片段在大化学库里的出现频次。常见,加分;少见,扣分。
最终的 SA Score 就是两项加起来,映射到 1 到 10 分。1 分意思是极易合成,10 分意思极难合成。这么一来,不同分子的合成难度,就能直接横向对比了。
这个模型算得飞快,给一个分子打分,毫秒级就能出结果。模型逻辑说得清,药物化学家一看就懂,也愿意用。不用复杂的算力支持,随便就能部署。
但短板也很突出。它分不太清 “结构复杂” 和 “合成困难”,有些分子看着结构绕,但其实有成熟的合成路线,它很可能会误判,把合成难度估高了。而且它的评分只看静态的结构特征,根本没考虑化学反应到底能不能走通。
BR-SA Score:给 SA Score 打个补丁
BR-SA Score 基本保留了 SA Score 的框架,但它精准补上了 SA Score 关键短板 —— 没考虑化学反应的实际可行性。
它最核心的升级是碎片来源的评分机制。它把 “商品化砌块库里的片段” 和 “靠化学反应生成的片段” 分开,把专业逆合成软件 CASP 里的反应规则、砌块库这些核心知识,直接编码成可计算的化学指纹分数,相当于做了一个 CASP 的轻量级替代模型。这么一改,打出来的分,更接近一个老合成人员的直觉,而不只是看静态结构。
SYBA:贝叶斯那套概率游戏
SYBA(SYnthetic Bayesian Accessibility),是基于贝叶斯概率理论做的合成可及性预测模型。
它就回答一个很朴素的问题:一个分子的局部片段,在容易合成的分子里出现多,还是在难合成的分子里出现多?
它用贝叶斯算概率。统计 ECFP4 分子指纹片段,在易合成分子库和难合成分子库里的出现频率,再根据片段的频率差异,判断这个分子到底是易合成还是难合成。模型会给每个片段算一个 “易合成倾向性”,最后把概率整合起来,就得到了整个分子的合成可及性判断。
好处是算法易于理解,你能看见每个片段的贡献——哪个地方拖了后腿,一目了然,便于理解决策依据。
短板也有。它默认各个片段之间是相互独立的,忽略了片段之间形成化学键的实际难度。而且它用来训练的难合成分子样本,是靠算法生成的,样本好不好,直接决定了模型准不准。
SC Score:从反应数据里偷师
SC Score(Synthetic Complexity Score)是个分水岭。它不靠专家打分,而是从大规模真实反应数据里找规律。
它只认一条铁律,产物的复杂度,一定是大于等于反应物的复杂度。就靠这个 “方向性约束”,摸透什么结构容易获得,什么结构难合成。
训练的时候,它让神经网络给任意一组反应对(反应物→产物)打分,必须满足 “产物的分数≥反应物的分数 + 固定差值”。这么一搞,SC Score 学的就不是“复杂度指标”,而是“合成路径里复杂度是单向增加的”这个化学常识。
它最厉害的地方,不是又做了一个分子复杂度的指标,而是真正学会了 “合成就是让结构变复杂” 这个最基础的化学规律。所以它的评分,天然就贴合真实的合成逻辑,不是只看分子的静态结构。
RA score:逆合成规划的快速替代
如果对几百万甚至数千万个分子做虚拟筛选,每个都跑完整逆合成分析,电费账单分分钟教做人。
这时,RA score(Retrosynthesis Accessibility Score)出现了,它是专门为了效率优化做的二分类模型,最核心的作用就是快速判断一个分子能不能用专业逆合成软件 AiZynthFinder,规划出完整的逆合成路径。
好处很直接,比跑完整的逆合成分析快太多,完全能满足高通量筛选的需求,部署也简单,不用搭复杂的逆合成引擎。
但它只能给 “是 / 否” 的定性判断,给不出合成难度的连续量化分数。而且它的能力上限,完全取决于底层CASP 工具的水平。
DeepSA:拿 NLP 的方法读分子
Deep SA 算是把自然语言处理 (NLP) 技术,跨界用到了化学领域。它把分子的 SMILES 字符串当句子处理,套用 NLP 里的预训练语言模型,直接吐出来“容易合成”或“难合成”的标签,同时可输出相应的分类置信度。
这里要提一句,它的训练标签来自 Retro* 这类软件的回溯路径步数 ——10 步以内算易合成,超过 10 步或者规划失败就算难合成,不是用真实的实验数据训练的。
所以它学的其实是 “算法能不能规划出来”,不是 “实验室里能不能真的合成出来”,实际用的时候,一定要注意这个差别。
GASA:用图神经网络找关键子结构
GASA(Graph Attention-based Synthetic Accessibility),把分子建模为原子-键图,原子是节点,化学键是边,用多头图注意力层,自动学习关键的结构特征,还融合了键级信息,强化全局的表征能力。
图注意力机制,能让模型自动把注意力放在影响合成难度的关键子结构上。
它的泛化能力强,能敏锐捕捉到结构上的微小变化带来的合成难度巨大差异,哪怕两个分子结构长得很像,合成难度天差地别,它也能分得清清楚楚。而且可解释性很好,能做到原子级的贡献可视化,直接给你看哪些原子、哪些子结构,对合成难度的影响最大,化学家用起来也省心。
当然也有短板。它的训练标签,同样依赖 Retro * 这类逆合成工具,和真实的合成情况,难免会有偏差。另外,图注意力的计算量比传统指纹模型大,超大规模筛选用它得掂量一下效率。
ChemAIRS 极速模式:基于真实合成步数的探索
聊完这些主流模型,再说说智化科技的创新方案—— ChemAIRS 极速模式。它直接用真实的合成步数,来做合成可及性评估,在很多地方都做出了突破。
先说说它和其他模型不一样的地方。
它完全靠真实世界的数据驱动。直接用 150 万 + 药化分子的真实最短合成步数,来做模型的训练和测试,既不依赖人工定的复杂规则,也不靠逆合成软件的间接预测。就靠真实的实验数据建模,自然更贴合实验室里的真实合成场景。
它用了很严谨的时间分割验证。2023 年之前的数据,用来训练和验证,2023 年之后的数据,单独拿来做测试。这种评估方式,能真实看出模型对全新类药分子的泛化评估能力,不会因为数据泄露,把模型的性能估高了。
它能自主学习非线性关系。用神经网络做端到端的学习,自己就能抓住分子结构和合成步数之间,复杂的非线性关系。和传统方法不一样,ChemAIRS 极速模式输出的,是和合成步数高度相关的连续评分,不是简单的 “能 / 不能” 的二分类,给药物化学家的决策依据,要精细得多。
它整合了千万级的先验知识。模型前期就整合了千万级的已知分子和商品化原料信息,能很好地解决常规模型的一个通病 —— 对 “看着结构复杂,但其实有成熟合成路线” 的分子,经常出现误判。同时评估速度也很快,完全能满足高通量筛选的需求。
模型整体性能对比
我们在 2023 年之后的独立测试集上,给这些主流模型做了系统性的评估。
结果显示,ChemAIRS 极速模式的预测值,和真实合成步数的皮尔森相关系数是最高的,展现出卓越的预测准确性。SC Score 和经典SA Score 紧随其后,表现也不错。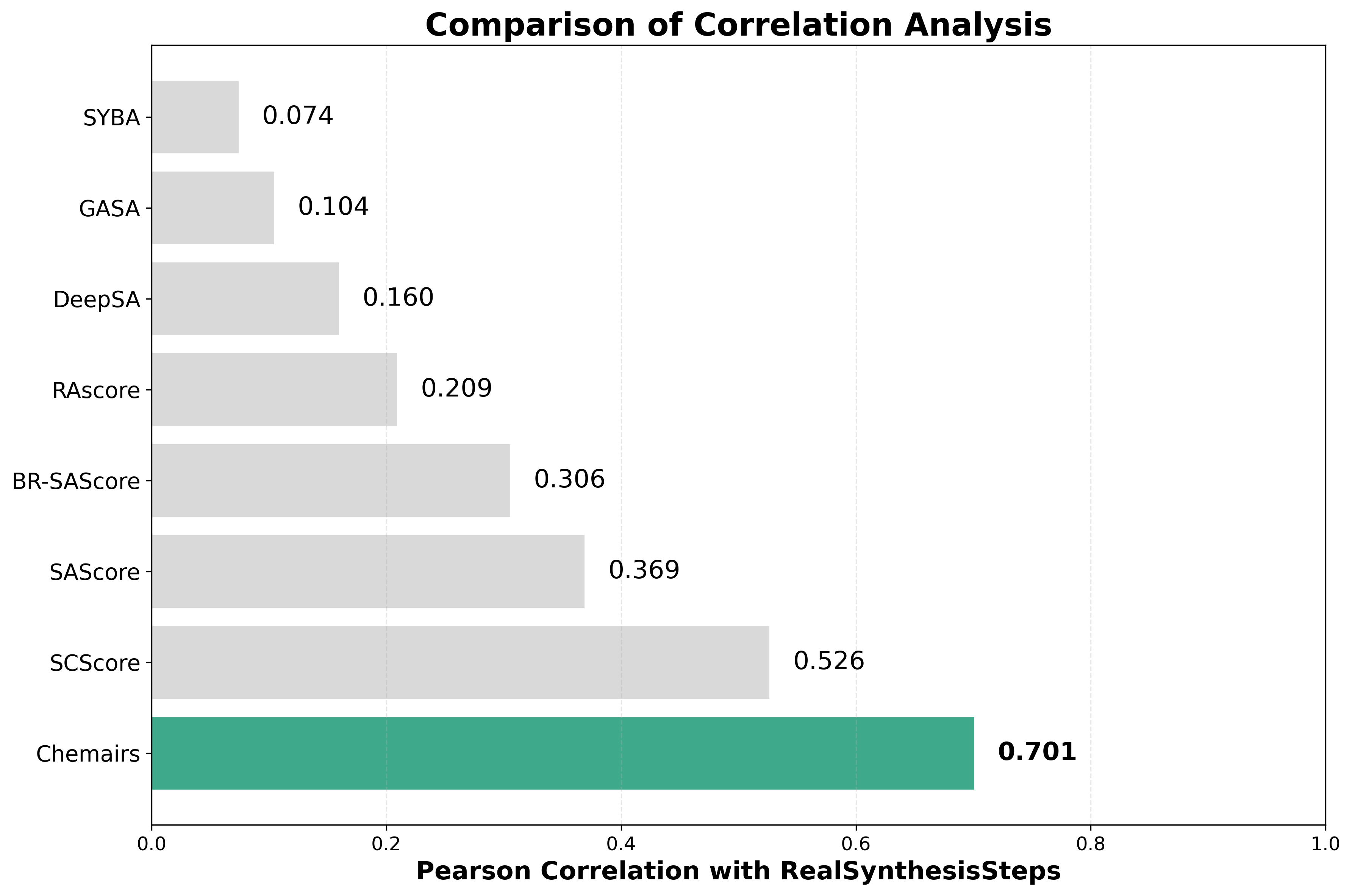
图1. 各模型预测值和真实值皮尔森相关系数对比
虚拟筛选场景性能对比
再看更贴近实际应用的虚拟筛选场景。我们筛选 2 万个分子做后续的深入分析,结果:
- ChemAIRS:16,400 个分子能够被成功合成
- SCScore:14,000 个分子能够被成功合成
- SAScore:1,200 个分子能够被成功合成
- 随机选择:仅 6,200 个分子能够被成功合成
ChemAIRS 不光在 Top20K 的筛选精度上最高,性能稳定性也很好。随着采样分子数量的增加,它的预测精度是缓慢下降的,不会出现断崖式的跌落。这个特性,对真实的药物研发项目来说,价值非常大。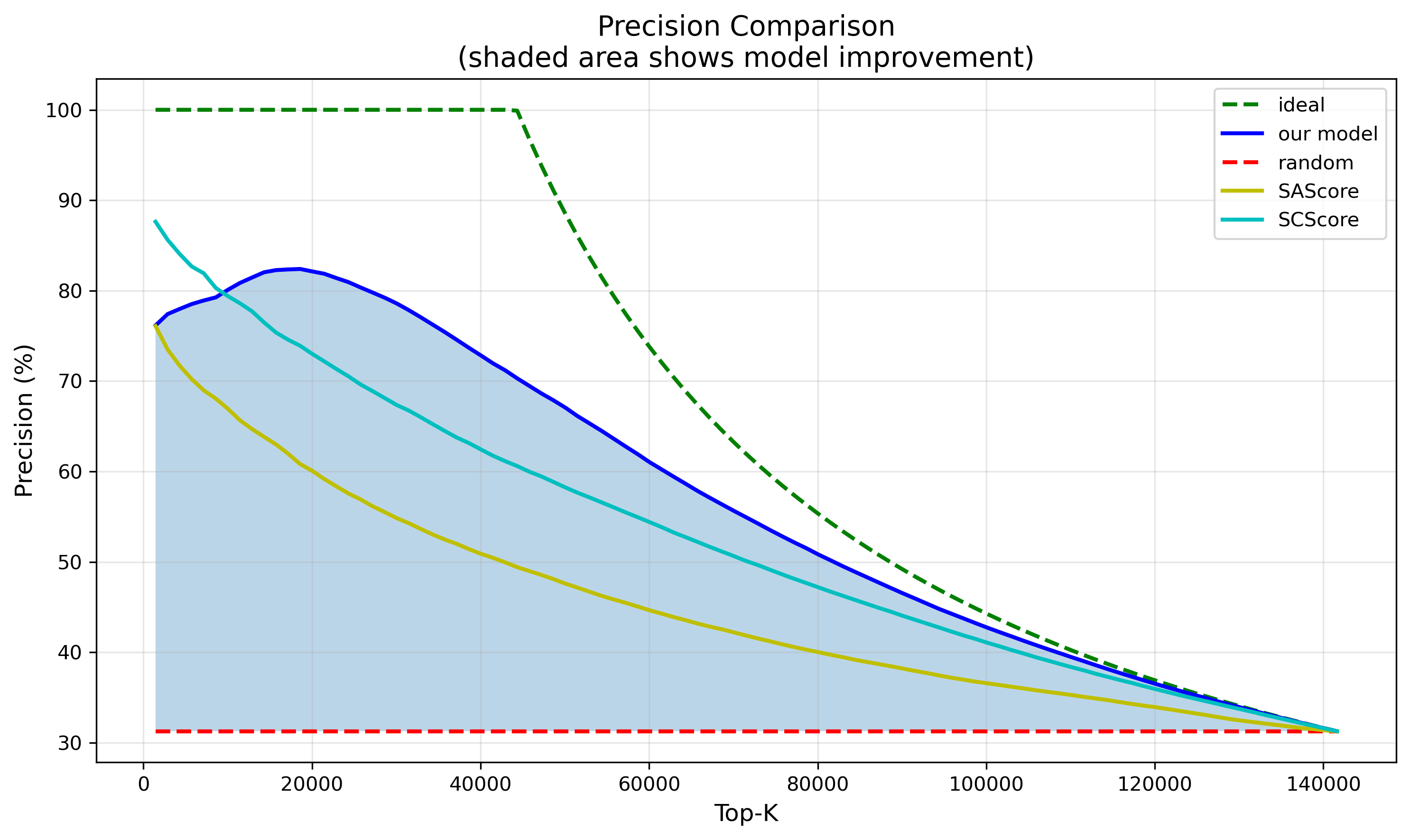
图2. 模型对测试集中前 10% 分子的预测精度变化对比
从 SA Score 开创的规则化方法,到 SC Score 带起来的数据驱动路线,再到 ChemAIRS 靠真实合成步数做的创新探索,这十几年合成可及性评分一直在进化。
ChemAIRS 的极速模式,评估 100 万个分子仅需约半小时,能快速把易合成的分子筛出来。但得承认,0.7的相关系数不是满分,有些分子看似好合成,在实际操作中可能还会面临挑战。
所以,对准确性和可解释性要求更高的场景,更推荐用 ChemAIRS 的其他模式。这些模式基于逆合成推理,能给出更精准的合成可行性评估,可解释性也更强,还能给药物化学家提供具体的合成建议和路线参考。
合成可及性评估技术的每一次进步都在一点点打通 AI 分子生成和实验合成之间的壁垒,推动药物研发往更高效、更智能的方向走。