ChemAIRS 为手性四氢吡咯设计合成路线
手性胺是药物分子的“黄金骨架”,畅销药中超过 40% 含有这类关键结构。然而,传统外消旋体拆分法效率低下,浪费严重。如何高效、精准地构建手性中心,尤其是复杂杂环胺(如手性四氢吡咯),一直是合成化学的挑战。
已上市药物分子 Larotrectinib 有一单元结构为手性四氢吡咯衍生物。文献报道了多种手性合成策略,包括亚胺的酶催化还原和不对称还原。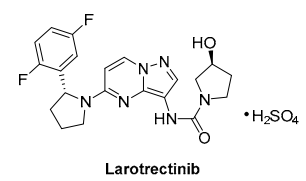
ChemAIRS 为目标分子合成提出了 5 条路线,供科研工作者参考。
路线一
通过廉价的原料 1a 制备格式试剂后,与 Boc 保护的吡咯烷酮反应得到中间体 2a,脱 Boc 保护形成内亚胺,然后经过亚胺的不对称还原得到手性中间体,再脱保护得到目标化合物。共四步反应,可行性高,其中不对称还原反应有文献成熟可靠的方法作为参考。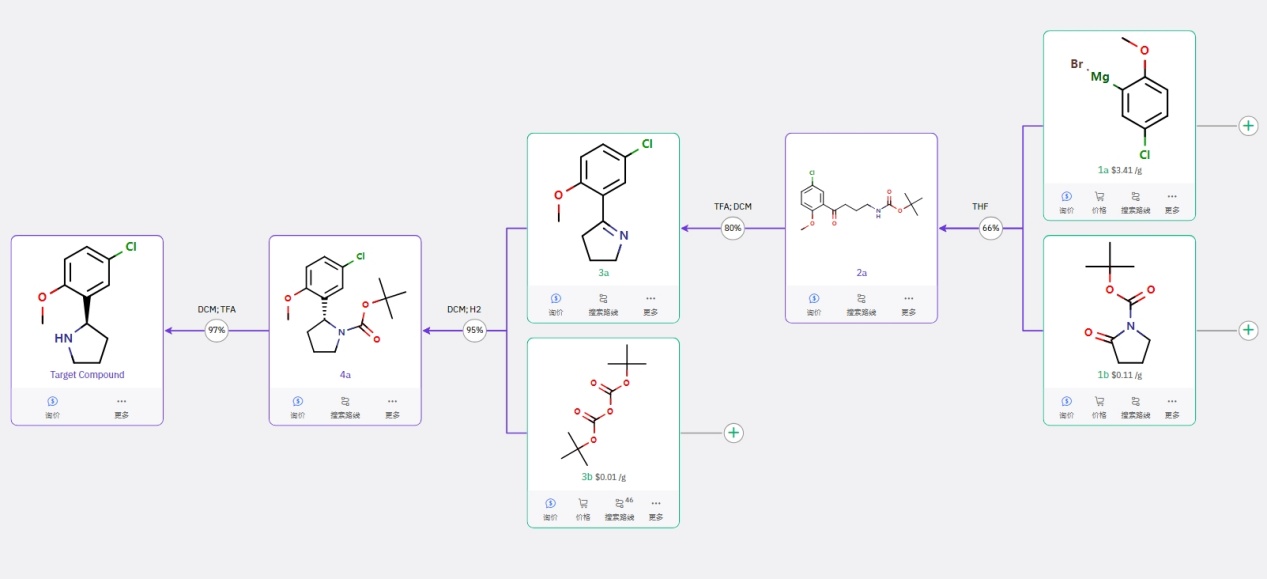
如文献报道了内亚胺在手性钌催化剂下不对称氢化的研究,ee 值可高达98% [1]。
路线二
1a 经傅克酰基化反应得到 2a,经转氨酶催化反应得到手性胺,该手性合成方法成熟可靠,适合放大。进一步进行分子内关环得到 4a,还原酰胺得到目标产物。共四步反应,手性构建策略可行性高,其中酮羰基的酶催化合成手性胺的策略可进行工业化放大。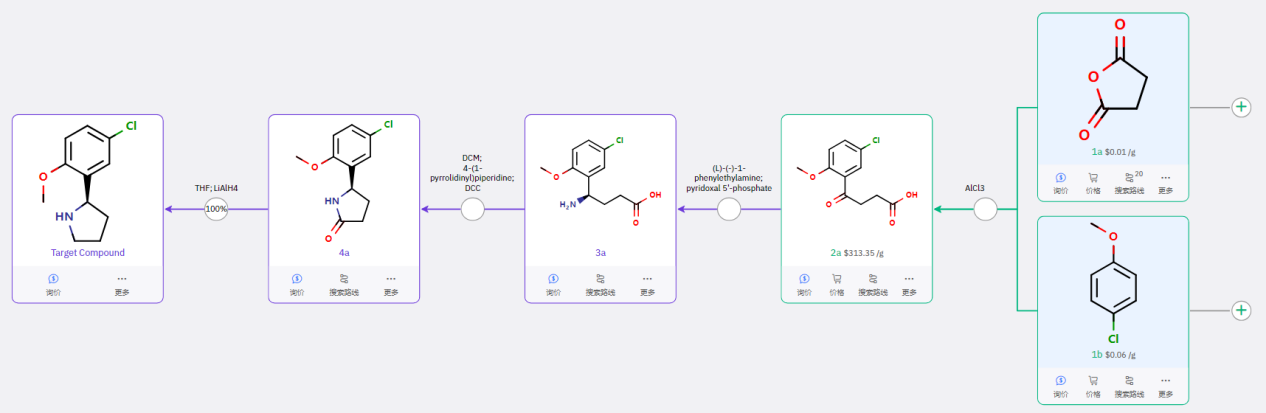
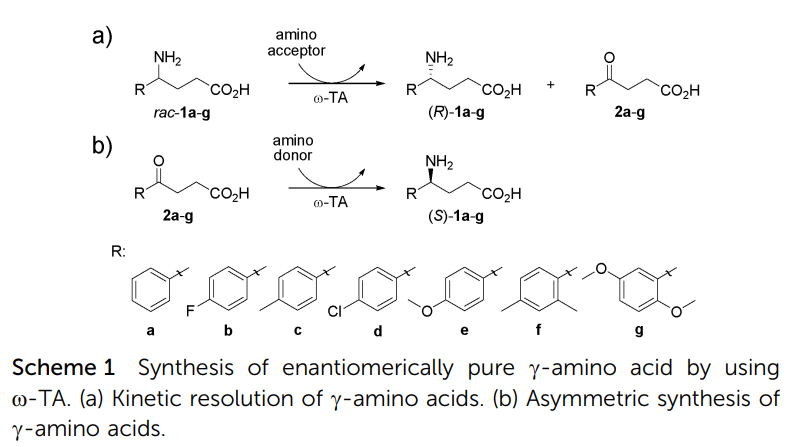
手性构建步骤 [2]
路线三
该路线手性引入策略简单可行,唯一不足是用到强碱叔丁基锂,安全风险较高,同时也用到了贵金属催化剂,适合进行小规模制备。路线通过 1a 与 1b 在强碱、路易斯酸及手性配体,在钯催化剂条件下完成两个片段的不对称偶联,最后脱保护得到目标化合物。两步反应,手性构建策略有较高的合成难度,不对称合成反应有已知文献作为参考。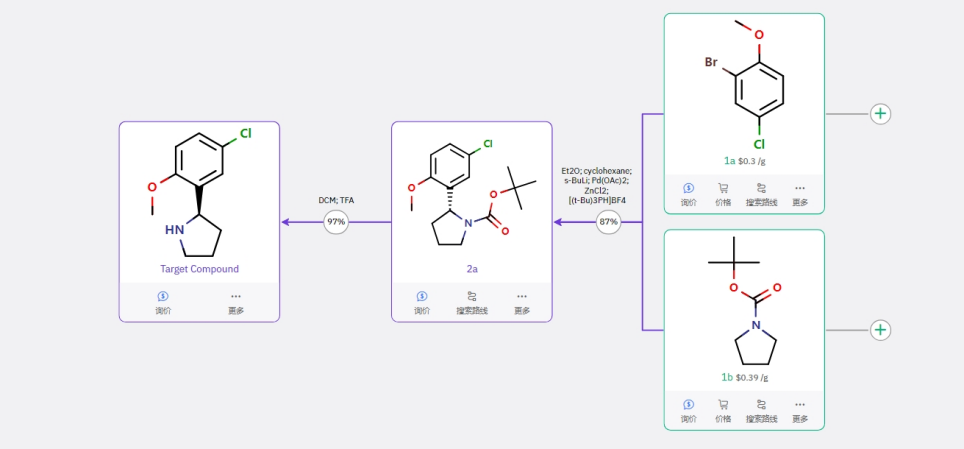
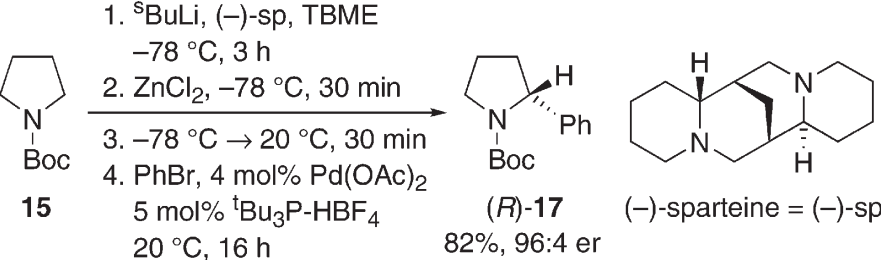
手性构建步骤 [3]
路线四
该路线手性的引入策略较为新颖,风险较高,但仍然值得尝试。通过 1a 水解得到 2a,与 2b 制备的格氏试剂进行亲核加成得到 3a,氰基经还原得到胺基,通过羟基与胺基基不对称的烷基化得到目标产物。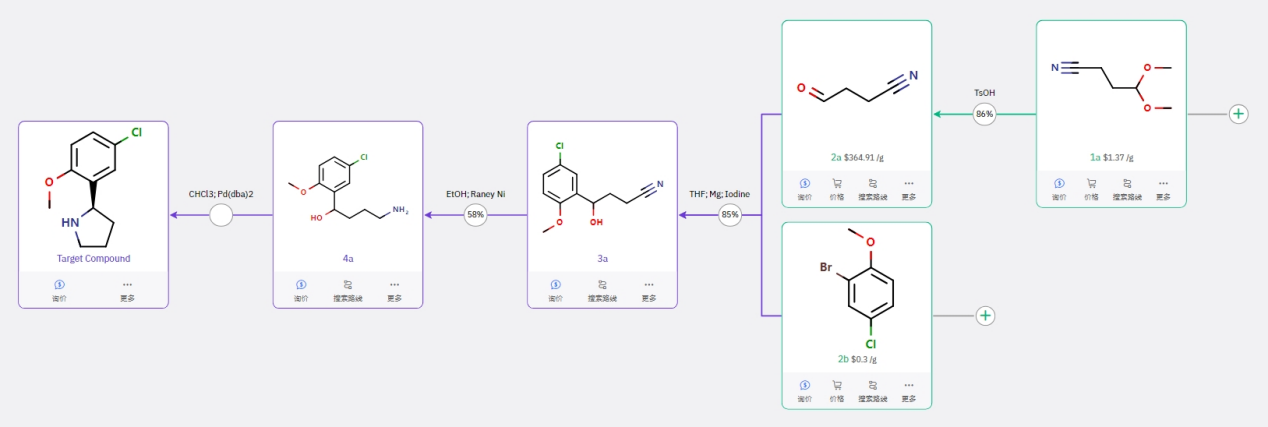
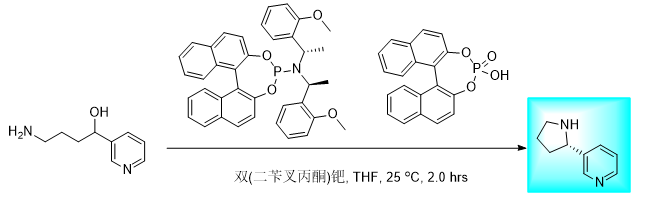
路线五
该路线手性引入策略较为新颖,风险较高,但仍然值得尝试。通过 1a 和 1b 经不对称脱羧偶联得到手性中间体,再进行脱保护即为目标产物。该策略基于文献中已经成熟的脱羧偶联而提出的,而目前最前沿的不对称合成策略中,可用消旋的原料来完成手性单元的构建 [4]。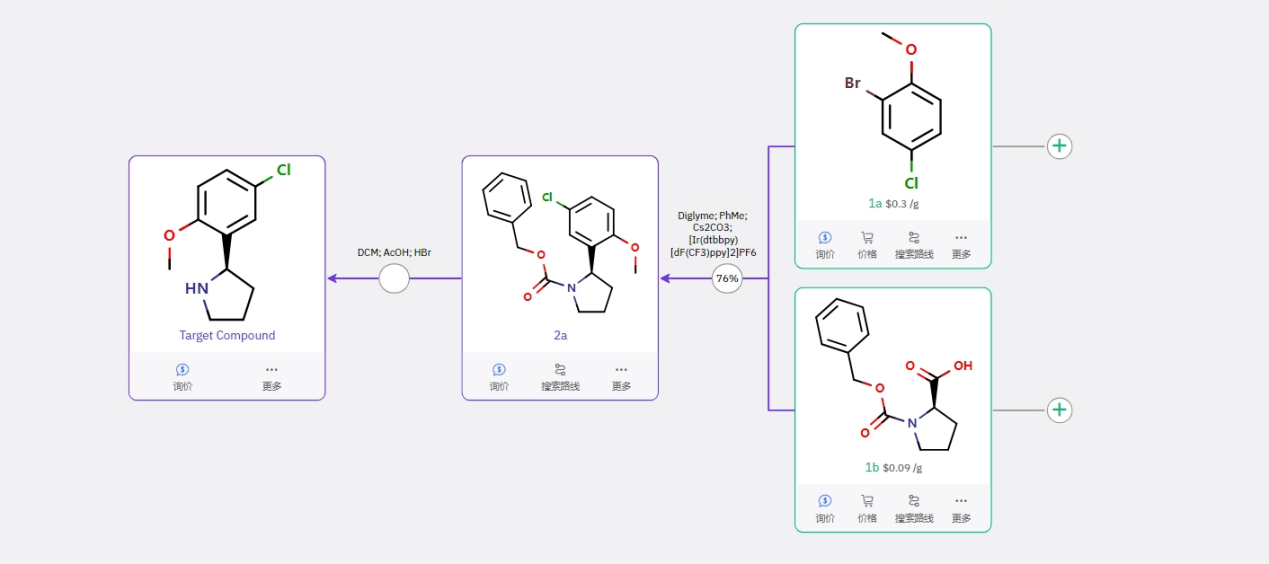
文献中的反应条件如下: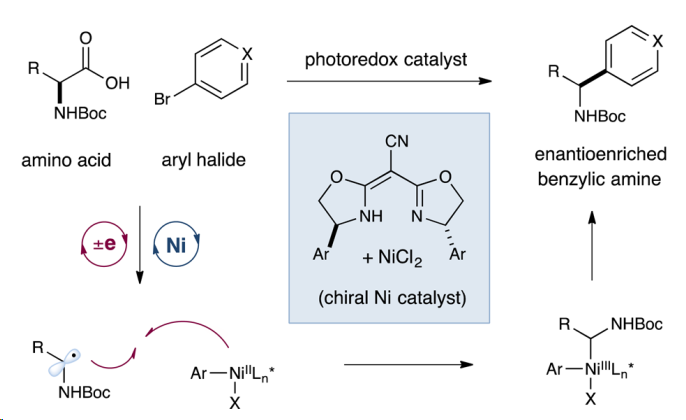
从上述的路线分析可以看出,ChemAIRS 基于数据学习和挖掘,能利用已知或类似文献报道的方法提出稳健的手性构建策略。同时,也能根据现今科研前沿,提出较为新颖的合成方案。虽然部分方案从反应的机理来说具有较大的风险,但仍然不失为一种值得研究的思路。作为逆合成路线设计工具,ChemAIRS 在手性化合物的路线设计上同样表现优越,为科研人员快速精准找到实施方案提供了重要手段。
参考文献:
1. Chen F, Ding Z, Qin J, et al. Highly effective asymmetric hydrogenation of cyclic N-alkyl imines with chiral cationic Ru-MsDPEN catalysts[J]. Organic Letters, 2011, 13(16): 4348-4351.
2. Shon M, Shanmugavel R, Shin G, et al. Enzymatic synthesis of chiral γ-amino acids using ω-transaminase[J]. Chemical Communications, 2014, 50(84): 12680-12683.
3. Barker G, et al. Enantioselective, palladium-catalyzed α-arylation of N-Boc pyrrolidine: in situ react IR spectroscopic monitoring, scope, and synthetic applications. J Org Chem. 2011 Aug 5;76(15):5936-53.
4. Zuo, Zhiwei, et al. "Enantioselective decarboxylative arylation of α-amino acids via the merger of photoredox and nickel catalysis." Journal of the American Chemical Society 138.6 (2016): 1832-1835.




